




Exp 006
 Objective:
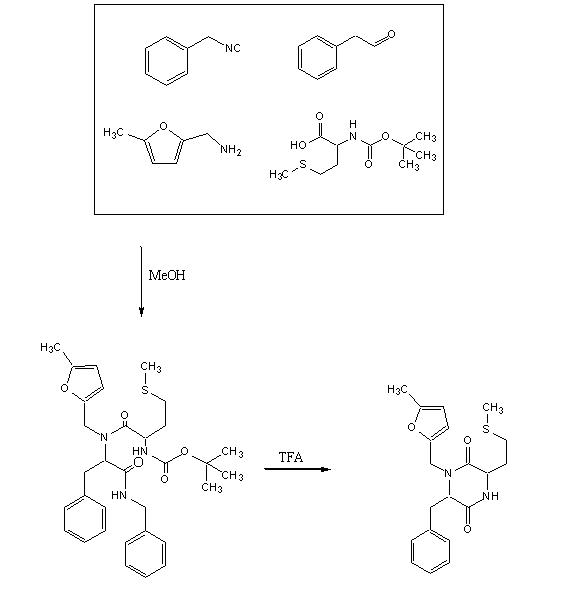
Objective:To test out the Ugi Synthesis and cyclization to a diketopiperazine using phenylacetaldehyde, 5-methylfurfurylamine, N-(tert) butoxycarbonyl)L-methionine, and benzylisocyanide using the protocol described here. The target diketopiperazine is not predicted to be active but is a close analog of the product that we wish to make once the catechol aldehyde is obtained.
As pointed in one of the comments posted to exp 003, the complete reaction was carried out at a higher concentration for each of the component involved. Also, the reaction mixture was heated to 80C for 30 min in 1,2-dichloroethane and TFA at the uncyclized stage.
Procedure:
To a 50 ml Erlenmeyer flask was added methanol (20 ml), phenyl acetaldehyde (55µl, 0.49 mmol) , 5-methylfurfurylamine (55µl, 0.49 mmol) , benzylisocyanide 60µl, 0.49mmol) and N-tert-butoxycarbonyl L-methionine (124.7 mg, 0.50 mmol). The mixture was stirred for 24 h, evaporated, refluxed for 30 min in 1,2-dichloroethane (9 ml) and trifluoroacetic acid (1 ml) then evaporated again to a dark oil. The crude product was taken up in dichloromethane (5 mL), washed with 5% HCl, dried over anhydrous MgSO4 and evaporated to yield a dark red oil.
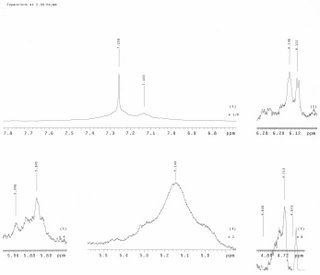
H-NMR of the crude product in CDCl3:

 TLC after extraction using 1:1 5%HCl/DCM
TLC after extraction using 1:1 5%HCl/DCM
Discussion
The H-NMR of the crude product does not show an aldehyde peak in the 9-10 ppm region, indicating that at least one starting material is consumed. The crude product may contain both cyclized a diketopiperazine and deprotected uncyclized amine but the NMR of the mixture cannot distinguish between these. For a possible mechanism of the Ugi reaction, click here.
Conclusion
This experiment will have to be aborted if the HNMR obtained is to be trusted. It can be seen that the furan peaks at 5.9 ppm are not strong enough to advocate an isolation and purification.
This experiment helped me acquaint with the chromatotron, NMR and refreshed other experimental techniques .
Log
- 8:38 pm (03/28/2006): In a 50ml Erlenmeyer flask, 20ml of methanol placed. To it 55µl (0.49mmol) of phenyl acetaldehyde, 55µl (0.49mmol) of 5-methylfurfurylamine, 60µl (0.49mmol) of benzylisocyanide and 0.12468 gms (0.50mmol) of N-(tert) butoxycarbonyl) L-methionine was added. An FTIR spectrum was obtained at t=0.
- The reaction was stirred at room temperature with a teflon coated stir bar on a stir plate.
- 8:38pm (03/29/2006): Stirring was stopped.
- 8:45pm (03/29/2006): Methanol was evaporated from the sample using a rotovap (75 C).
- 10:30pm (03/29/2006) Added 9ml of 1,2-dichloroethane and 1ml of trifluoro acetic acid to the methanol void sample in a round bottom flask. Heated the reaction mixture for about 5 mins before the vapors got intense and had to attach a reflux condenser, the heating was turned off after 30 mins..; the boiling points of the solvents are, TFA= 72.4 ° C and 1,2-DCE = 83 °C
- 11:10pm (03/29/2006) After 30 mins the solvents were evaporated using a rotovap, at 85C.
- 11:40pm (03/29/2006) The residue is a dark brown liquid.
- 12:10am (03/29/2006) A TLC of the product was run in a mixture of 10ml 1,2-dichloroethane and 10ml of hexanes.
- H-NMR of the crude final product was also obtained. (Varian 300Mz Instrument)
- 2:30pm (04/02/2006) Extraction: Added 5ml of 5% HCl and 5 ml methylenechloride to the round bottom flask containg the crude product and transferred it to a separatory funnel. Separated the bottom methylenechloride layer from the top aqueous layer. Washed the aquoues acid layer twice with 5mls of methylenechloride and collected the bottom methylenechloride layer each time. Combined the organic layers and dried it over anhydrous magnesium sulfate (MgSO4). Evaporated the dichloromethane using a rotovap and collected the extracted product. A TLC of the product was obtained (Shown above).
- 11:30 am (05/10/2006) Separation using a Chromatotron : The compound was dissolved in 10 ml of methylene chloride, applied to the rotating plate on the chromatotron.The round bottom flask was rinsed with 1ml of methylene chloride, the residue was poured over the rotating plate of the chromatotron. A mix of 1:1 hexanes and methylene chloride was used initially as the mobile phase. (60mls).
In the mean time fractions were collected. To speed-up the movement of the compound a more polar solvent mix was used. The composition of the mobile phase was changed from 1:1 hexanes, methylene chloride to 1:1 methylene chloride, methanol (gradually), and finally to methanol.
posted by Khalid Mirza @ 3/29/2006 02:44:00 PM
1 comments
![]()

1 Comments:
Matt -
1) thanks - we'll fix this shortly
2) Yes we'll get a diastereomeric mixture from this reaction. Actually Find-A-Drug did not provide stereochemical info in the library they sent us. If any of these are active in the anti-malarial test we'll separate them.
3) thanks!
Post a Comment
<< Home