




Exp 014
The Ugi Rerun of experiment 006
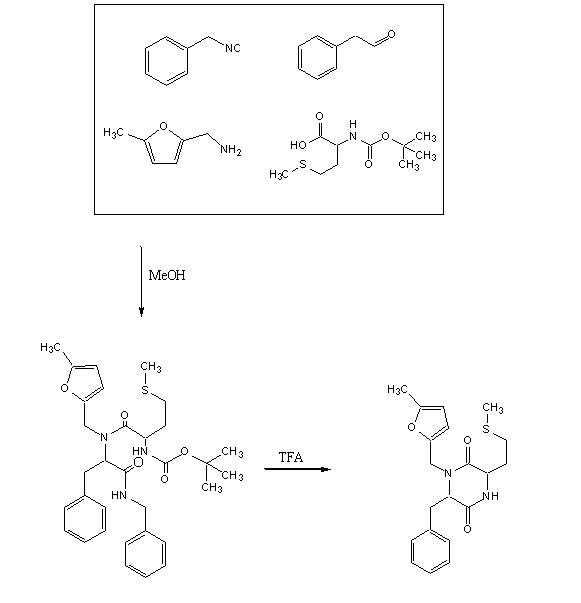
Objective : To accomplish the Ugi Synthesis and cyclization to a diketopiperazine using phenylacetaldehyde, 5-methylfurfurylamine, N-(tert) butoxycarbonyl)L-methionine, and benzylisocyanide using the protocol described here. The target diketopiperazine is not predicted to be active but is a close analog of the product that we wish to make once the catechol aldehyde is obtained.
Procedure:
To a 50 ml Erlenmeyer flask added methanol (20 ml), phenyl acetaldehyde, 623µl , 5-methylfurfurylamine 555µl, benzylisocyanide 608µl, and N-(tert) butoxycarbonyl) L-methionine 1.24604 gms. The mixture was stirred for 15 h, evaporated, refluxed for 45min in 1,2-dichloroethane (27 ml) and trifluoroacetic acid (3 ml) then evaporated again to a dark oil. The crude product was taken up in dichloromethane (30 ml), washed with water, dried over anhydrous MgSO4 and evaporated again. A dark, reddish oil was obtained.
Log
1. 9:00pm (05/30/2006) In a 50ml Erlenmeyer flask, 20ml of methanol placed. To it 623µl of phenyl acetaldehyde, 555µl of 5-methylfurfurylamine, 608µl of benzylisocyanide and
1.24604 gms of N-(tert) butoxycarbonyl) L-methionine was added.
2.The reaction was stirred at room temperature with a teflon coated stir bar on a stir plate for 15 hours (stopped stirring at 12:00 noon (05/31/2006). (Videos of the reaction being stirred)
3.Methanol was evaporated on a rotovap set at 100C, Obtained 3.239gms the crude product.
4. Added 27ml of 1,2-dichloroethane and 3ml of trifluoro acetic acid to the methanol void sample in a pre weighed round bottom flask. Refluxed the contents of the flask for 45 mins (the rheostat settings had to be adjusted because the reaction mixture has stopped refluxing during the process),the boiling points of the solvents are, TFA= 72.4 ° C and 1,2-Dichloroethane = 83 °C
5.The reaction mixture was rotovaped (set at 100C) again after 45mins on reflux.
6.The resultant compound was washed with water (twice, 70mls and 30 mls) and partitioned in methylene chloride (60mls)
7.After separation; the organic layer was dried over anhydrous Magnesium Sulfate, filtered.
8. The obtained crude product was subjected to an NMR analysis.
9.2:30pm (06/01/2006) The crude product was further dried under vacuum until it foamed and settled. The process was continued for a period of one and half hours.(4:00pm)
9. After evaporation under vacuum 3.5174gms of product was obtained.
10. 1.3051 gms of the product was weighed and intorduced on a chromtotron. Intially a 2:1 mix of hexanes and methylene chloride was used (about 400ml), 19 of 20mls portions were acuumulated.
11. Most of the compund stillremained on the rotor disc. The compound was let dry
overnight on the rotor.
Upon Dr. Bradley's suggestion the remaining compound was removed from the rotor using methanol.
12. The compound in methanol was rotovaped and evaporated under low pressure, and introduced back on the chromatotron, however the the eluent used this time was 1:1 hexanes
and methylene chloride (100mls mix) The eluent composition was changed gradually from 1:1 hexanes, methylene chloride to just methylene chloride (100mls), then it was further changed to 0.5% methanol in methylene chloride.
Polarity of the solvent was changed regularly. 0.5% methanol, 1% methanol, 2% methanol, 3% methanol,
4% methanol, 6% methanol, and 8% methanol in methylene chloride.
16 fractions were accumulated.
16 fractions were accumulated.
posted by Khalid Mirza @ 6/01/2006 05:08:00 PM
0 comments
![]()

0 Comments:
Post a Comment
<< Home