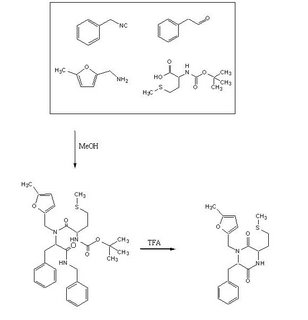
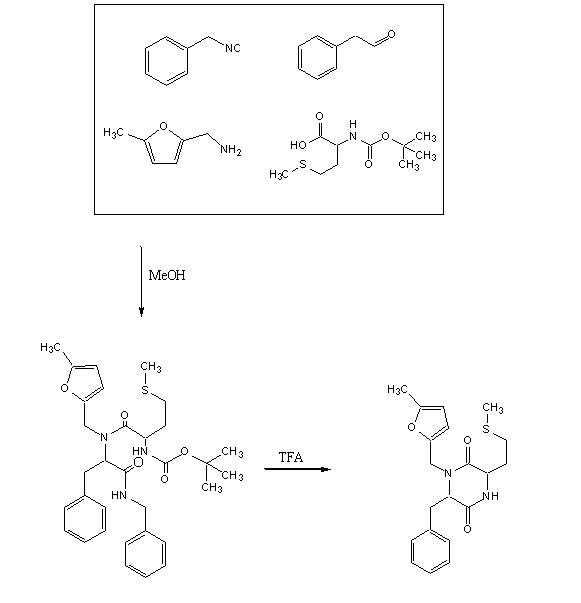
 Objective
Objective:
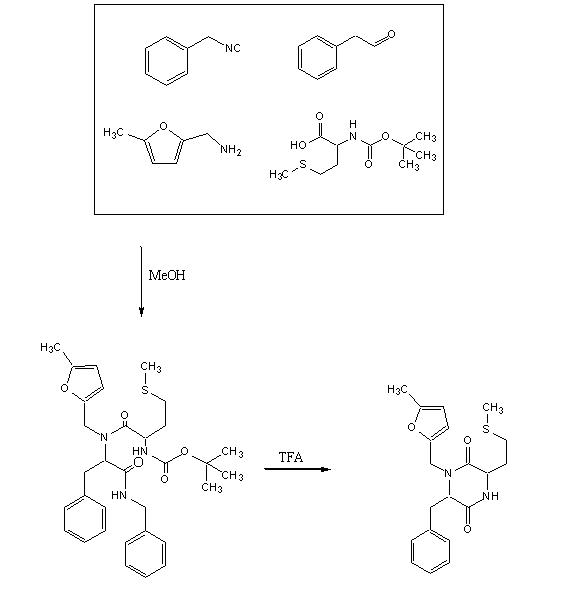
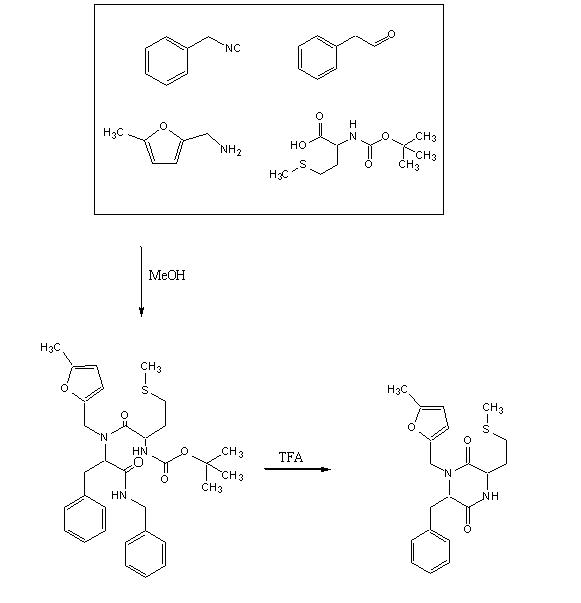
To test out the Ugi Synthesis and cyclization to a diketopiperazine using phenylacetaldehyde, methyl furfurylamine, N(tert-Butoxycarbonyl)L-methionine, and benzylisocyanide using the protocol described
here. The
target diketopiperazine is not predicted to be active but is a close analog of the
product that we wish to make once the
catechol aldehyde is obtained. 2006-02-09
Procedure PartA:
Reagents in MeOHPlaced 50mL methanol in an 125mL Erlenmeyer flask. Added 23 microliters
phenylacetaldehyde(0.21mmol), 24 microliters
5-methylfurfurylamine(0.21mmol, 52.66mg
N-(tert) butoxycarbonyl)L-methionine (0.21mmol), and 26 microliters of
benzylisocyanide (0.21mmol) in that specific order. Solution was flushed with nitrogen and stoppered. The reaction was stirred at room temperature ovenight. Various samples were taken out over time to monitor the reaction via TLC.
Results of PartAmass spec are inconclusive. See discussion for detailed explanation.
Procedure PartB:
TFA AdditionPippetted out 5mL of reaction solution. Evaporated solvent in vaccuo at 65 degrees Celsius. Prepared solution of 10% TFA in methylene chloride. Added 10mL of TFA solution to products.
Results of PartB2006-03-10
Mass Spectrum from 2006-02-17 Mass Spectrum of the
matrix used in MS MS of 3H MS of 3G MS of 3IDiscussionBased on the results of the TLC we can postulate that the intermediate diketopiperazine is there, but not the cyclized version. This is evidenced by the TLC plates where we were expecting the final product to have moved faster with the eluent. I believe the product didn't cyclize was because it was not heated in the last step.
Concerning the FAB-MS analysis of this experiment, it does not look like anything useful can be extracted. None of the pure starting materials show M+ peaks, just a lot of matrix peaks. Also, it is unclear that the calibration is correct based on a possible assignment of a strong peak corresponding to a benzyl fragment at 91.6 (off by 0.6 amu)
phenylacetaldehyde expect M+ at 120 but don't see that in either spectrum (
Mass Spectrum1,
Mass Spectrum2) - in both spectra see peak at 91.6 (is this benzyl fragment off by 0.6 amu?)
5-methylfurfurylamine expecting MH+ at 116 but don't see any there or nearby (
Mass Spectrum)
N-(tert) butoxycarbonyl)L-methionine expecting M+ at 249 - there is a small peak at 251.0 (
Mass Spectrum)
benzylisocyanide expecting M+ at 117 but nothing nearby (
Mass Spectrum) - note that phenyl isocyanide is drawn on this spectrum ConclusionWithout better testing this experiment will have to be done again. NMR and MALD/I is suggested.
LOGPartA13:03 Solution was flushed with nitrogen and stoppered. 2mL of starting solution was placed in a 1 dram vial. (sample 3A)
13:50 2mL of the reaction was pippetted out and placed in a 1 dram vial. (sample 3B).
14:15 2mL of the reaction was pippetted out and placed in a 1 dram vial. (sample 3C).
15:05 2mL of the reaction was pippetted out and placed in a 1 dram vial. (sample 3D).
17:05 2mL of the reaction was pippetted out and placed in a 1 dram vial. (sample 3E). Let reaction continue to stir over night.
2006-2-13
Reaction continued to stir over the weekend. 11:00 2mL of reaction was pippetted out and placed in a 1 dram vial (sample 3F) TLC plates of samples 3B, 3C, 3D, and 3E were run against the starting sample (3A) using methanol as the eluent. All of the samples (including 3A) had an Rf value of approximately1.
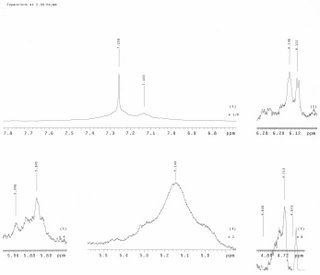
A TLC was run in 4:1 Hexanes and Dichloromethane with 3A as the standard against the final sample of the Ugi synthesis. The Rf value of the sample is 0.35 while the standard is 0.08. IR graphs were made for the Ugi synthesis at t=o, 50min., 72min, and 120min. 30microliter samples of the Ugi synthesis were evaporated on a KBr pellet. Graphs of the KBr pellets were also made to determine any possible error. It was determined by the t=72min graph that there was an insufficient amount of sample to provide a clear graph and all that was seen was the vibration of the material. The t=120min had a sample of 60microliters which produced a better plot.
2006-02-14 (James)
Submitted a 0.5mL sample of the reaction mixture (now 5 days at room temp) for
MS analysis by FAB. The product mass weight was found with very little of the reactants. However the peak of this is small, and there is an overwhelming peak of a mass of 436.7. What this is, is not yet known. The sample that was given was not heated in TFA (trifluoroacetic acid) so we are not yet sure that this is the cyclized diketopiperazine. This is the
uncyclized product. Also the 436.7 peak could have something to do with the golden yellow color that we have seen in the samples.
2006-02-16 (James)
11:00. Solutions of the reactants of the mock Ugi synthesis were prepared to perform a TLC vs the 3F sample of the reaction (which was taken two days after the solution was heated) in order to determine if the reactants are still present. Solutions were made as follows in 2mL glass viles: (16A) 30microliters of Phenylacetaldehyde in 1mL MeOH. (16B) 30microliters of Methylfurfurylamine in 1mL MeOH. (16C) 30microliters of Benzylisocyanide in 30mL MeOH 11:45. The results of the TLC in 4:1 hexanes and methylenechloride are as follows: (16A) The Rf value of the standard (3F) was 0.32, while the reactant was 0.23 (16B) The Rf value of 3F was 0.38 while the reactant did not travel at all. (16C) The Rf value of 3F and the reactant was 0.21. They traveled approximately the same distance.
12:15. A solution of the last reactant was made to do a TLC. (16D) 0.0011g of N-(tert-Butoxycarbonyl) in 1mL MeOH.
12:30. Results of the TLC in 4:1 hexanes and methylenechloride are an Rf value of 0.32 for the standard while the reactant did not move.
13:10. The reactant solutions (16A-D) were given over for MS analysis along with a sample of t=0 of the mock Ugi reaction and a sample of the extraction from 13t[2] (See
Exp 002). The t=0 solution was was golden yellow as the rest of the samples taken from this reaction.
PartB
2006-02-17 (Alicia)
Phenylacetaldehyde (
Mass Spectrum1,
Mass Spectrum2) Methyl furfurylamine (
Mass Spectrum) N(tert-Butoxycarbonyl)L-methionine (
Mass Spectrum) Benzylisocyanide (
Mass Spectrum)
15:02 Solution turned from bright yellow to deep red-brown. Pippetted out approximately 1 mL and placed in a 1dram vial (sample 3G). Allow solution to stir at room temperature for 1 hr. Ran a TLC plate of sample 3G against our original starting solution (3A) using 1:1 methylene chloride/hexane. The starting sample had an rf value of approximately 0.2 while 3G had an rf=0.
16:02 Pippetted 1mL of reaction and placed in a 1 dram vial (sample 3H, t=1hr). Ran TLC plate against sample 3A. Evaporated remaining solution in vaccuo at 65 degrees Celsius and dissolved product in methylene chloride(sample 3I). A 0.5mL sample of 3G, 3H, and 3I will be submitted for MS on Monday.
2006-02-20
Samples were submitted for MS. IR graphs were also mad with sample
3G and
3I.
2006-03-02 (Alicia)Ran TLC of phenalacetaldehyde against samples 3B, 3C, 3D, 3E, 3F, 3G, and 3H. Ran TLC of 5-methylfurfurylamine against samples 3B, 3C, 3D, 3E, 3F, 3G, and 3H. The eluent used for both was 9:1 hexane/methylene chloride. The 5-methylfurfurylamine appeared to run down into the eluent, will re-run TLC. It was determined that these samples were overloaded. Will re-run samples of 3F(right before TFA soln was added) and 3H (after TFA was added).Will upload pictures of the TLC plates as soon as equipment becomes available.
2006-03-08 (Alicia)
Prepared a phenylacetaldehyde/methanol solution by adding 4.6uL phenylacetaldehyde to 10mL of methanol. Ran a TLC plate against samples 3F and 3H using a 6:4 methylene chloride/hexane solution as the eluent. The picture may be somewhat unclear. Prepared a 5-methylfurfurylamine/methanol solution by adding 4.8uL 5-methylfurfurylamine to 10mL of methanol. Ran a TLC plate against samples 3F and 3H using a 6:4 methylene chloride/hexane solution as the eluent. Prepared a N(tert-butoxycarbonyl)L-methionine/methanol solution by adding 5.3mg N(tert-butoxycarbonyl)L-methionine to 5mL methanol. Ran a TLC plate against samples 3F and 3H using a 6:4 methylene chloride/hexane solution as the eluent. Prepared a benzylisocyanide/methanol solution by adding 5.2uL of benzylisocyanide to 10mL of methanol. Ran a TLC plate against samples 3F and 3H using a 6:4 methylene chloride/hexane solution as the eluent. These numbers were chosen to reflect the concentration of the reactants in the original starting solution.
2006-03-09
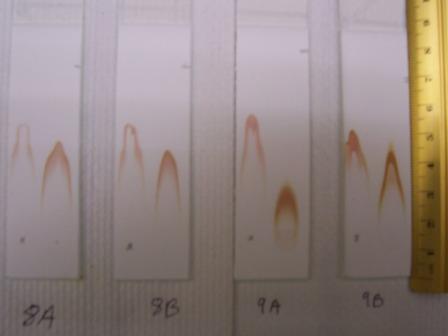
Made dilute samples of the raw starting materials for TLC. Phenylacetaldehyde (18P), Benzylisocyanide (18B), and Methylfurfurylamine (18M) where made as 30microliters samples in 1 mL methanol while a small amount of crystals was used to make the sample of N-(tertButoxcarbonyl)-L-methionine (18N) in 1mL methanol. The TLC was don in 1:1 hexanes methylenechloride. TLC plates are marked as the samples were named. The spots on the TLC plate from left to right are the sample, after TFA, and before TFA.
TLC Pic1 TLC Pic22006-03-14 (Alicia)
Based on previous TLC of the reaction it was concluded that we have obtained the linear intermediate. More TFA will be added to the solution and then refluxed. TLC of the reaction will be taken to see if the reaction cyclized. This will be evidenced by a faster moving spot than the linear intermediate. Prepared a 10% TFA/methylene chloride solution. Added 20mL TFA solution to products (3I). Attached flask to a reflux condenser and ran under nitrogen gas. Took sample of the solution (3J). Turned on heat at 12:19.
12:40 Reaction began refluxing.13:09 Took sample of the refluxing reaction (3K). Ran TLC against 3J using 1:1 methylene chloride/hexanes as the eluent.
13:50 Took sample of the refluxing reaction (3L). Ran TLC against 3J using 1:1 methylene chloride/hexanes as the eluent.
14:35 Took sample of the refluxing reaction (3M). Ran TLC against 3J using 1:1 methylene chloride/hexanes.17:00 Took sample of the refluxing reaction(3N). Ran TLC against 3J using 1:1 methylene chloride/hexanes. The TLCs don't appear to have any spots on them. The amount of product that was left in the flask was uncertain which may explain why there aren't any moving spots on the TLC plates. It may be worthwhile to restart this experiment using dichloroethane as the solvent instead of methylene chloride. Aborted.
NEXT-submit reactants and reaction before and after TFA for usual MS analysis (not FAB)-update discussion for TLC analysis and future TLC work -determine if the reaction worked and if not, what happened











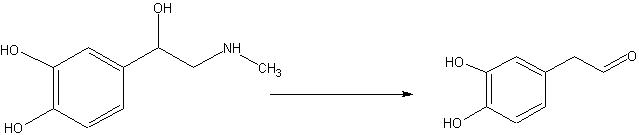


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}